Alpha Thalassemia được xem là một trong những nguyên nhân hàng đầu dẫn tới tình trạng thiếu máu và tan máu ở trẻ em. Nguyên nhân gây ra tình trạng này là do suy giảm hoặc thiếu hụt tổng hợp chuỗi Alpha Goblin trong phân tử Hemoglobin. Bệnh có thể tồn tại dưới dạng thể nhẹ không cần điều trị hoặc thể nặng buộc phải chăm sóc y tế suốt đời.
Bài viết này được viết dưới sự hướng dẫn chuyên môn của các bác sỹ thuộc Trung tâm Công nghệ cao Vinmec.
1. Định nghĩa và phân loại bệnh Alpha Thalassemia
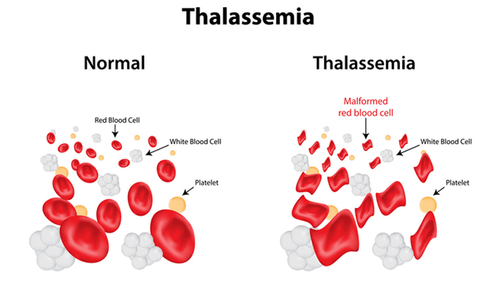
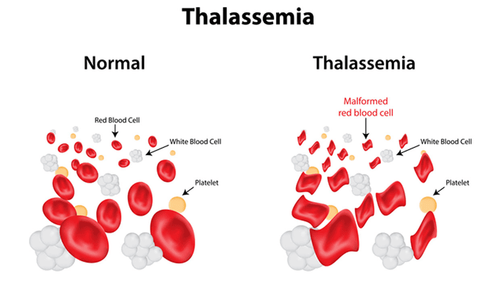
Mỗi người đều có các gen để tạo ra huyết sắc tố trong những tế bào hồng cầu giúp vận chuyển oxy trong cơ thể. Tuy vậy, một số trường hợp lượng huyết sắc tố được tạo ra ít hơn bình thường, đó là khi mang các đặc điểm của bệnh Alpha Thalassemia (Tan máu bẩm sinh thể Alpha). Đây là một bệnh di truyền lặn có đặc trưng gây suy giảm hoặc thiếu hụt tổng hợp chuỗi Alpha Globin trong phân tử Hemoglobin. Lý do cho điều này là việc có ít hơn 4 bản sao gen Alpha-Globin. Hiện nay, bệnh Alpha Thalassemia được phân thành 8 loại bệnh phổ biến.

1.1. Gen Triplicated Alpha
Đây là nhóm đặc điểm của những người có nhiều hơn 4 gen Alpha-Globin, chỉ có thể phát hiện thông qua thử nghiệm di truyền. Các bản sao này mang đặc điểm Beta-Thalassemia dẫn đến tình trạng thiếu máu nghiêm trọng cần được truyền máu.
1.2. Silent Carrier State (Thể Ẩn)
Người mang mầm bệnh này chỉ có 1 gen Alpha-Globin bị ảnh hưởng, đặc điểm nhận biết là tế bào hồng cầu bình thường hoặc hơi nhỏ, chỉ có thể xác nhận bằng xét nghiệm di truyền. Mặc dù không cần điều trị nhưng khả năng di truyền lại cho các thế hệ con cái là khá cao.
1.3. Alpha Thalassemia Trait
Đây là khi có hai gen Alpha-Globin bị ảnh hưởng, đặc điểm nhận biết là các tế bào hồng cầu nhỏ không đáp ứng với chất bổ sung sắt và bị thiếu máu nhẹ không cần điều trị.
1.4. Bệnh Hb H
Đối với trường hợp này chỉ có 1 gen Alpha-Globin hoạt động, dẫn đến tình trạng thiếu máu nhẹ đến trung bình, cần tới xét nghiệm di truyền để phát hiện. Những người mắc bệnh này có khả năng cần được điều trị và phải tránh dùng một số loại thuốc nhất định.
1.5. Hb Constant Spring (Hb CS)
Tình trạng được xác định khi một gen Alpha-Globin tạo ra một loại huyết sắc tố khác không hoạt động tốt với những thay đổi nhỏ về hồng cầu hoặc thậm chí là không có. Thể bệnh Alpha Thalassemia này cũng không cần điều trị và có thể phát hiện thông qua xét nghiệm di truyền.
1.6. Homozygous Hb Constant Spring
Tình trạng này được xác định thông qua đặc điểm có hai gen Alpha-Globin tạo ra Hb Constant Spring và hai gen hoạt động bình thường, dẫn tới thiếu máu nhẹ, có thể khiến cho lá lách hoặc gan to hơn. Cần điều trị nếu xuất hiện các triệu chứng thiếu máu trầm trọng, đặc biệt đối với thai nhi.
1.7. Bệnh Hb H-Constant Spring
Bệnh Hb H-Constant Spring là khi có cả hai đặc điểm Alpha Thalassemia lẫn Hb CS, chỉ có một gen Alpha-Globin hoạt động, với mức độ thiếu máu từ trung bình đến nghiêm trọng. Bệnh lý này nên được điều trị và tránh dùng một số loại thuốc, đặc biệt với thai nhi vì có khả năng xuất hiện các dấu hiệu thiếu máu nghiêm trọng.
1.8. Alpha Thalassemia Major
Đây được xem là tình trạng nghiêm trọng nhất ở trẻ em, chỉ có thể xảy ra khi cả cha và mẹ đều mang các đặc điểm của bệnh tan máu bẩm sinh thể Alpha. Bệnh có thể gây ra những vấn đề nghiêm trọng khi mang thai khiến người mẹ tử vong và sẽ gây tử vong cho thai nhi nếu không được điều trị.

2. Nguyên nhân và triệu chứng bệnh Alpha Thalassemia
Nguyên nhân chủ yếu của tình trạng này là sự đột biến (hoặc thay đổi) gen kiểm soát lượng Alpha-Globin được tạo ra, gây ra sự mất cân bằng so với Beta-Goblin, từ đó dẫn đến sự thiếu máu và các vấn đề y tế khác có thể gặp phải. Người mắc bệnh có thể không có triệu chứng gì nghiêm trọng, chỉ bị mất máu nhẹ hoặc gặp phải một số triệu chứng đáng chú ý như thường xuyên ở trong tình trạng mệt mỏi, tim đập nhanh hay bị hụt hơi và da nhợt nhạt.
Bên cạnh đó, bệnh nhân xuất hiện các dấu hiệu như vàng da vàng mắt, dậy thì muộn, thường xuyên ủ rũ, cáu kỉnh, có hình dạng xương mặt và xương đầu bị biến đổi. Đặc biệt đa số sẽ kèm theo tình trạng dư thừa sắt trong cơ thể, tổn thương chức năng của gan, tim và hệ thống nội tiết tố.
3. Các biến chứng có thể gặp phải khi mắc bệnh Alpha Thalassemia
Những người mắc bệnh Alpha Thalassemia có thể gặp phải nhiều biến chứng khá nghiêm trọng, ảnh hưởng đến sức khỏe như lá lách to vì thiếu máu, buộc bác sĩ phải cắt bỏ lá lách, từ đó làm tăng nguy cơ nhiễm khuẩn. Những thay đổi trong tủy xương làm cho xương bị biến dạng, dài hơn, nhỏ hơn và giòn hơn, do đó cũng dễ gãy hơn. Cuối cùng là nguy cơ tiểu đường do bị tổn thương hệ thống nội tiết và tuyến tụy.
4. Chẩn đoán và điều trị bệnh tan máu bẩm sinh thể Alpha
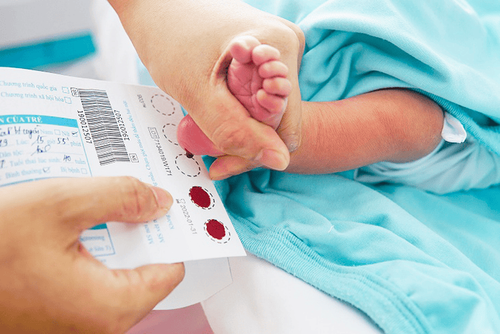
Xét nghiệm di truyền là một trong những biện pháp chẩn đoán xác định gen đột biến Alpha Thalassemia phổ biến nhất, thường được khuyến khích với các cặp đôi trước khi mang thai. Trẻ em sau sinh có thể lấy máu gót chân để sàng lọc bệnh, sau đó xét nghiệm gen chuyên sâu nếu phát hiện nguy cơ mắc bệnh liên quan đến Hemoglobin để hạn chế biến chứng. Các xét nghiệm máu thường được áp dụng khác bao gồm điện di huyết sắc tố và CBC (Công thức máu toàn phần).

Để đặt lịch khám tại viện, Quý khách vui lòng bấm số HOTLINE, đặt mua GÓI DỊCH VỤ hoặc đặt lịch trực tiếp TẠI ĐÂY. Tải và đặt lịch khám tự động trên ứng dụng My Vinmec để quản lý, theo dõi lịch và đặt hẹn mọi lúc mọi nơi ngay trên ứng dụng.